
Neurodegenerative Disorders
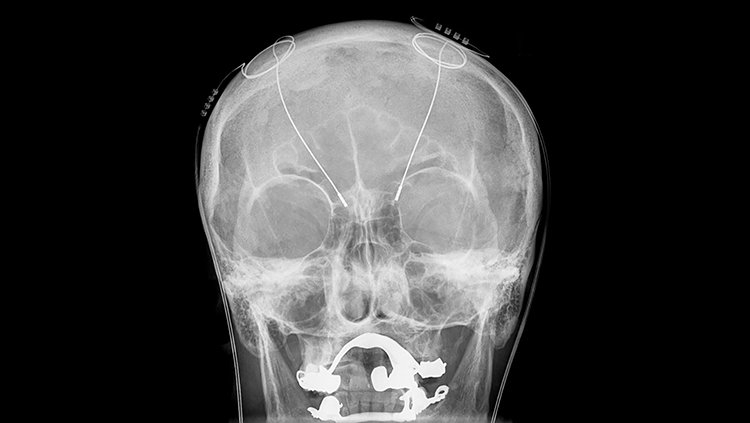
A new treatment for Parkinson's disease and other disorders arises from an outbreak of contaminated street drugs — and years of painstaking brain research.



