A History of the Classification of Glioma Brain Tumors
- Published9 Oct 2019
- Author Karen Hopkin
- Source BrainFacts/SfN
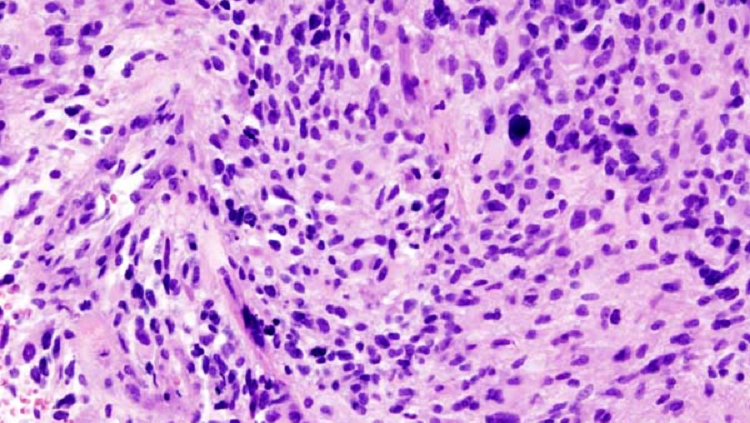
Classified
In 1918, American physician Percival Bailey had completed his medical internship and was anxiously pursuing his burgeoning interest in the workings of the brain. Hoping to continue his training, Bailey sent letters to two prominent members of the medical community: one, a psychiatrist; the other, a neurosurgeon. The neurosurgeon, Harvey Cushing, replied first, so Bailey joined him at the Brigham Hospital in Boston.
Cushing had been amassing an extensive repository of brain tumor specimens. At the time, doctors realized that some brain tumors grew aggressively, whereas others were relatively benign. But they had no way to know which was which. Bailey believed that Cushing’s collection might hold the clues to predicting how tumors would behave. He began by grouping the specimens according to the length of time each patient survived. He then exhaustively examined more than 400 tumors, correlating their microscopic appearance with clinical outcomes.
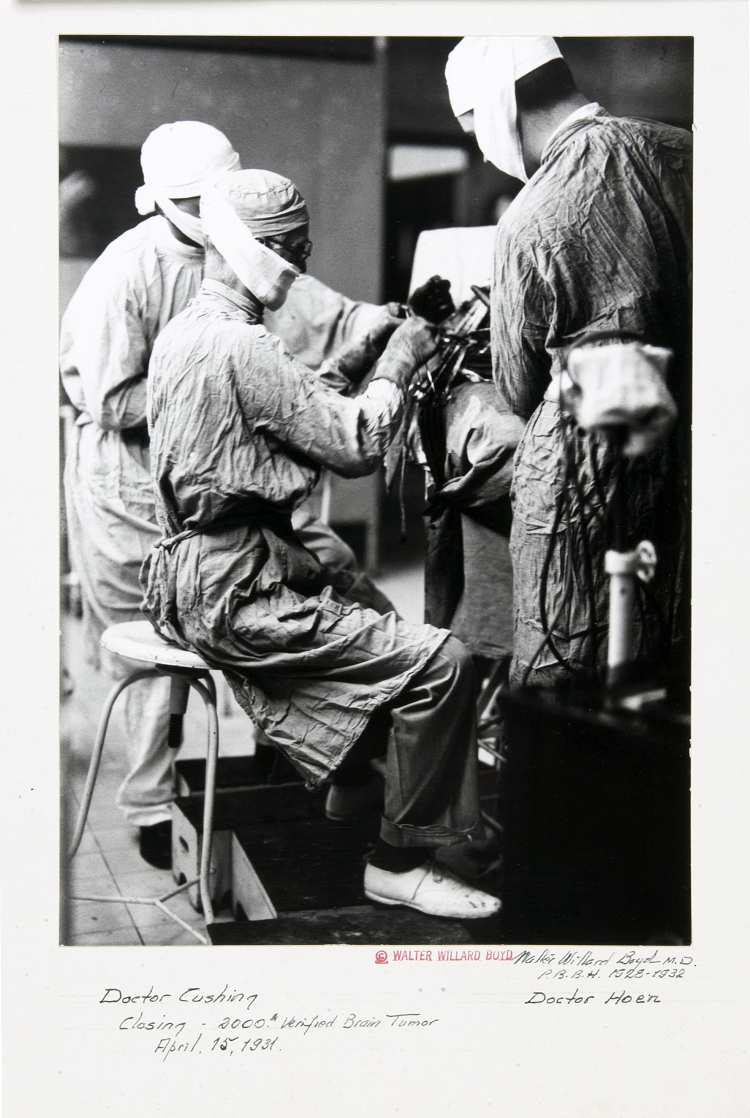
The resulting classification, published by Bailey and Cushing in 1925, demonstrated that the cellular structure of a tumor can guide treatment and prognosis. And it laid the groundwork for the system presented by the World Health Organization in 2016 to describe and diagnose gliomas: tumors that arise from glia, the various types of supportive cells of the central nervous system.
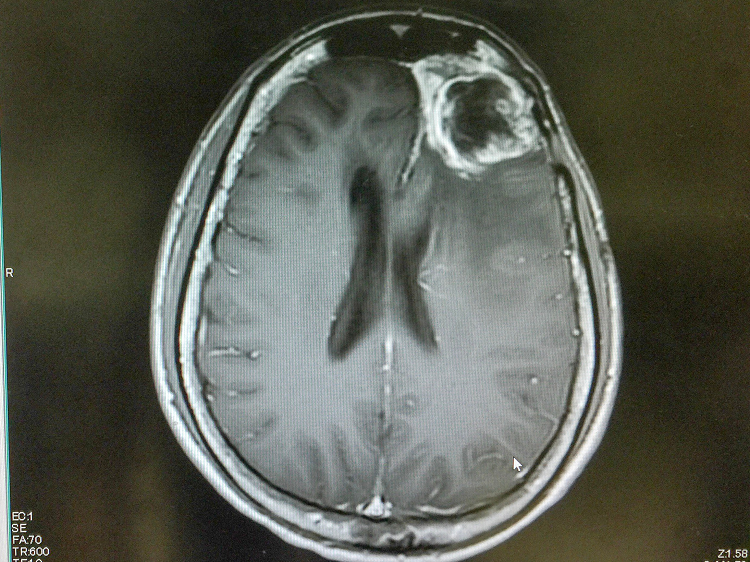
Glioblastomas — the most commonly diagnosed malignant brain tumor — constitute the most aggressive and lethal form of glioma. They are thought to arise from astrocytes, the star-shaped cells providing extensive physical and metabolic support for nerve cells, or their progenitors.
Glioblastomas are difficult to treat because of their location. “These tumors happen to occur in the most vital and complex organ in our bodies,” says Nduka Amankulor of the University of Pittsburgh Medical Center. “Removing these tumors surgically without damaging neurological function is challenging.”
Complicating matters, the cells in a glioblastoma behave like the parent cells from which they arise. Outnumbering neurons five-to-one, normal glia crawl along neural pathways to surround and entwine with their neuronal neighbors. This migratory behavior makes glioblastomas highly invasive and difficult to eradicate, as the tumors can extend microscopic tendrils stretching throughout the brain.
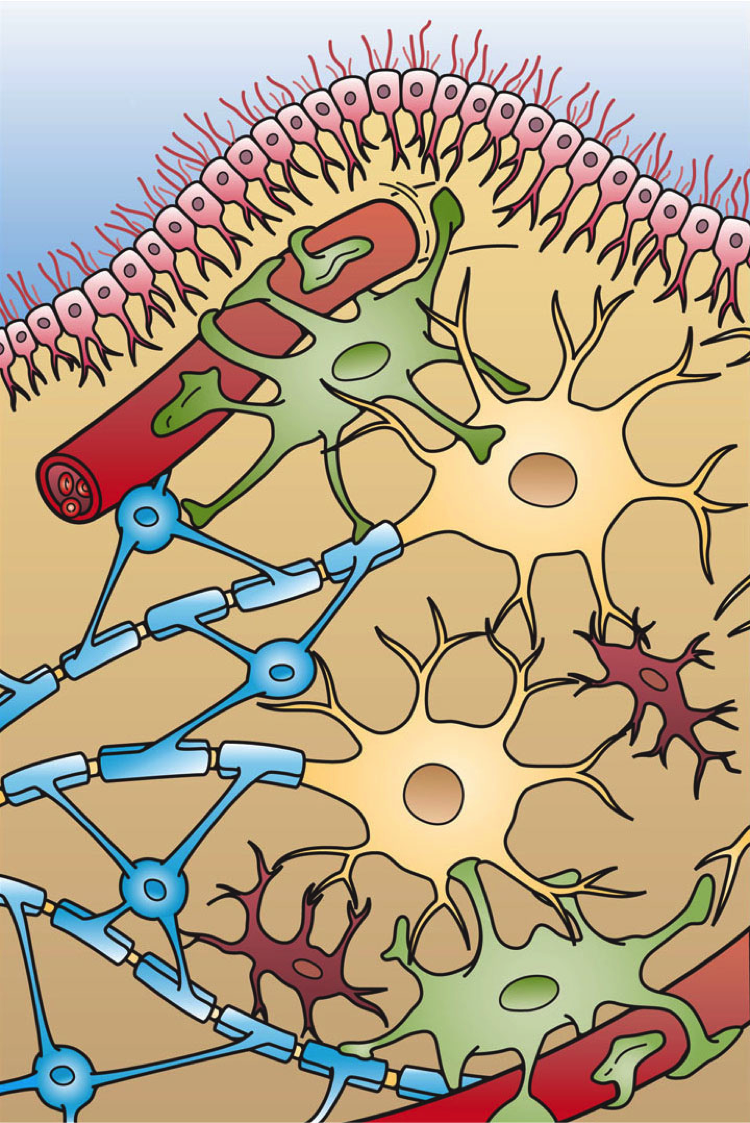
Molecular Blueprints
Modern genomic techniques are allowing researchers and clinicians to take Bailey and Cushing’s work to the next level: classifying glioblastomas at the molecular level. Such studies not only expand our understanding of what gives rise to these cancers, but also point toward more effective, targeted therapies.
In its pilot project, the Cancer Genome Atlas Research Network sequenced the DNA of glioblastomas donated by more than 200 patients. The results, published in 2008, highlighted the array of molecular derangements driving the growth of this aggressive cancer. Identifying the genes and pathways mutated in an individual glioblastoma provides clues to how aggressive a particular tumor is likely to be and how to select the best treatment options for each patient. For example, in a small subset of patients, an oncogene called Braf turns on inappropriately. These patients respond well to a class of drugs that specifically inhibit this overactive, mutant form of Braf.
A more common alteration involves a gene critical to DNA repair called MGMT (O6-methylguanine-DNA methyltransferase). About half of all glioblastomas inactivate MGMT, rendering those tumors inept at repairing DNA damage. The oral chemotherapy drug temozolomide can help these patients by causing DNA damage to build up in and incapacitate MGMT-deficient tumors.
For patients whose tumors possess active MGMT, temozolomide is less effective. Identifying this early can allow doctors to enroll them in clinical trials for drugs that work differently or to offer other treatment approaches.
Molecular profiling also sheds light on the mechanisms promoting tumor recurrence. Treatment of MGMT-deficient glioblastomas with temozolomide inevitably leads to the recurrence of tumors with an unusually large number of mutations — 50 or 60, rather than one or two. Such “hypermutable” tumors tend to be resistant to traditional chemotherapies. Temozolomide could be enhancing the formation of these tumors by inadvertently knocking out other key DNA repair pathways, resulting in the rapid accumulation of mutations.
Glioblastomas might best be treated by combining drugs that work like temozolomide with others that prevent the emergence of “hypermutable” tumors. Or by adopting treatments that take advantage of this genetic instability to wipe out recurring, secondary tumors.
Calling on Immunity
In the 1950s, average survival for patients with glioblastoma hovered around three months and many physicians chose not to treat. Average length of survival started to creep up as surgeons began to operate on the tumors. Now, the addition of chemotherapy and radiation to the surgical protocol have boosted survival rates to around 14 to 15 months — a marked improvement that researchers are working to extend much further.
Part of what makes glioblastomas so hard to treat is that the tumors, themselves, may be able to suppress natural immunity. Ashley Ghiaseddin of the University of Florida Health says this could open a treatment avenue. “So, the idea of immunotherapy is to wake up the immune system so it can attack tumors.”
Some cancers can thrive by making use of the kill switches on immune cells. Once activated, immune cells can be shut down again by tripping one of their built-in molecular kill switches, called checkpoints. Checkpoints prevent the immune system from attacking the body’s own tissues, and some cancers make use of them to hide from the immune system. Drugs called checkpoint inhibitors disable these natural brakes and rev up the immune response. They have shown promise in treating cancers like melanoma. Studies of checkpoint inhibitors in animal models of glioblastoma showed some benefit; however, studies in humans indicate checkpoint inhibitors may need to be combined with other treatments in concert with considering the molecular make of an individual patient’s tumor.
A 2017 clinical study pitting checkpoint inhibitors and the standard chemotherapy agent Avastin against Avastin alone did not show enhanced survival. Some researchers suggest this study failed to demonstrate any benefit because patients were accepted into the study irrespective of the molecular profile of their tumor. It is also possible that immunotherapies may be more effective in patients with hypermutable tumors because genetic instability can generate changes that can be picked up by the immune system: significantly altered DNA and even unusual protein antigens. Such never-before-seen, “non-self” molecules may signal to immune cells that the tumor cells are indeed foreign — and incite an attack. Case reports indicate patients with hypermutable tumors sometimes display dramatic responses to immunotherapy.
Researchers are also working to develop alternatives to traditional treatments like surgery and chemotherapy. Some are using devices to deliver electromagnetic pulses called tumor treatment fields to disrupt tumor cell division. A phase 3 clinical trial of this method, which offers a noninvasive option for patients who cannot tolerate surgery or whose tumors are inoperable, has shown positive results. Other researchers are deploying viruses — injected directly into the glioblastoma — to infect and kill tumor cells or to deliver molecules that can activate and attract immune cells. More than 20 percent of the patients who participated in a study conducted at Duke University starting in 2012 survived more than 3 years after diagnosis, and some are still alive today.
Advances in our understanding of the molecular drivers and signatures of individual tumors are transforming the way we treat glioblastomas. “What’s become apparent is that the one-size-fits-all approach that was used by the generation of oncologists and neurosurgeons before us is not the best way to treat this disease,” says Amankulor. “Now that we’ve entered the era of molecular medicine, the most effective approaches involve using a personalized treatment strategy perfectly tailored to each individual patient.”
About the Author

Karen Hopkin holds a PhD in biochemistry and is co-author of the textbook Essential Cell Biology.
CONTENT PROVIDED BY
BrainFacts/SfN
References
Cloughesy, T. F., Mochizuki, A. Y., Orpilla, J. R., Hugo, W., Lee, A. H., Davidson, T. B., … Prins, R. M. (2019). Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nature Medicine, 25(3), 477–486. doi: 10.1038/s41591-018-0337-7
Combs, S. E., Rieken, S., Wick, W., Abdollahi, A., von Deimling, A., Debus, J., & Hartmann, C. (2011). Prognostic significance of IDH-1 and MGMT in patients with glioblastoma: One step forward, and one step back? Radiation Oncology (London, England), 6, 115–115. doi: 10.1186/1748-717X-6-115
Daniel, P., Sabri, S., Chaddad, A., Meehan, B., Jean-Claude, B., Rak, J., & Abdulkarim, B. S. (2019). Temozolomide Induced Hypermutation in Glioma: Evolutionary Mechanisms and Therapeutic Opportunities. Frontiers in Oncology, 9, 41–41. doi: 10.3389/fonc.2019.00041
Ferguson, S., & Lesniak, M. S. (2005). Percival Bailey and the classification of brain tumors. Neurosurgical Focus FOC, 18(4). Retrieved from https://thejns.org/focus/view/journals/neurosurg-focus/18/4/foc.2005.18.4.8.xml
Filley, A. C., Henriquez, M., & Dey, M. (2017). Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget, 8(53), 91779–91794. doi: 10.18632/oncotarget.21586
Kaley, T., Touat, M., Subbiah, V., Hollebecque, A., Rodon, J., Lockhart, A. C., … Hyman, D. M. (2018). BRAF Inhibition in BRAFV600-Mutant Gliomas: Results From the VE-BASKET Study. Journal of Clinical Oncology, 36(35), 3477–3484. doi: 10.1200/JCO.2018.78.9990
Karachi, A., Dastmalchi, F., Mitchell, D. A., & Rahman, M. (2018). Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro-Oncology, 20(12), 1566–1572. doi: 10.1093/neuonc/noy072
Kazda, T., Dziacky, A., Burkon, P., Pospisil, P., Slavik, M., Rehak, Z., … Lakomy, R. (2018). Radiotherapy of Glioblastoma 15 Years after the Landmark Stupp’s Trial: More Controversies than Standards? Radiology and Oncology, 52(2), 121–128. doi: 10.2478/raon-2018-0023
Kessler, A. F., Frömbling, G. E., Gross, F., Hahn, M., Dzokou, W., Ernestus, R.-I., … Hagemann, C. (2018). Effects of tumor treating fields (TTFields) on glioblastoma cells are augmented by mitotic checkpoint inhibition. Cell Death Discovery, 4, 12–12. doi: 10.1038/s41420-018-0079-9
Killela, P. J., Pirozzi, C. J., Healy, P., Reitman, Z. J., Lipp, E., Rasheed, B. A., … Bigner, D. D. (2014). Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget, 5(6), 1515–1525. doi: 10.18632/oncotarget.1765
Louis, D. N., Perry, A., Reifenberger, G., von Deimling, A., Figarella-Branger, D., Cavenee, W. K., … Ellison, D. W. (2016). The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathologica, 131(6), 803–820. doi: 10.1007/s00401-016-1545-1
Marosi, C., & Preusser, M. (2017). Milestones of the last 10 years: CNS cancer. Memo, 10(1), 18–21. doi: 10.1007/s12254-016-0309-x
Preusser, M., Lim, M., Hafler, D. A., Reardon, D. A., & Sampson, J. H. (2015). Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nature Reviews. Neurology, 11(9), 504–514. doi: 10.1038/nrneurol.2015.139
Stoyanov, G. S., & Dzhenkov, D. L. (2018). On the Concepts and History of Glioblastoma Multiforme—Morphology, Genetics and Epigenetics. Folia Medica, 60(1). Retrieved from https://content.sciendo.com/view/journals/folmed/60/1/article-p48.xml
Cancer Genome Atlas Research Network. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455(7216), 1061–1068. doi: 10.1038/nature07385