Patient Driven: Neuroscientists Solve the Puzzle of the Gene-Protein Duo Behind SYNGAP1
- Published15 Oct 2019
- Author Charlie Wood
- Source BrainFacts/SfN
Most infants fuss and cry when they get sick, but Landen showed little sign of pain. Instead, when illness struck, Landen simply slowed down a bit.
“By the time I connected the dots and realized he was sleeping 15 minutes longer,” his mother Lisa says, “he’d have bilateral ear infections.”
In addition to his insensitivity to discomfort, Landen gets seizures, has an autism diagnosis, and at 12 years old knows about 100 words.
When he was born in 2006, Landen’s disorder had no name — just a constellation of symptoms that Lisa and her husband managed the best they could. It took genome sequencing in 2015 to match his condition to a recently discovered syndrome. Because of a small mutation in a gene called SYNGAP1 — possibly a single additional letter in the DNA code — Landen’s brain can’t make enough of a protein that keeps neural wiring in check as we grow and promotes learning and memory in adults.
It’s estimated that one percent of all intellectual disability not explained by a known syndrome is caused by these mutations.
As it turns out, Landen is not alone. Researchers now know of hundreds of children like him thanks to an online organization of affected families. “It’s estimated that one percent of all intellectual disability not explained by a known syndrome is caused by these mutations,” says Society for Neuroscience past president, Richard Huganir, a neuroscientist at Johns Hopkins University and co-discoverer of the gene-protein duo. “That’s lots and lots of kids.”
And as a result, neuroscientists like Huganir — who began their careers studying basic brain function — find themselves thrust toward the front lines of clinical research, turning neuroscience theory toward the search for treatments, and perhaps even cures.
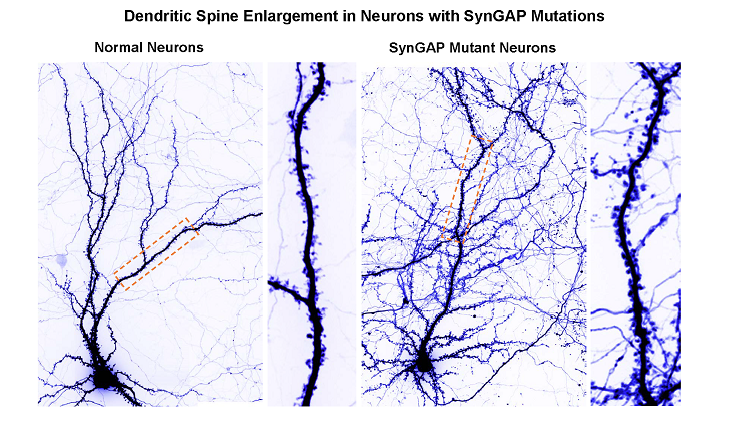
Mutations in the SYNGAP1 gene occur randomly in sperm or eggs, leaving developing brains with half the amount of the brain with half amount of a crucial protein it needs for normal development. This protein, called not surprisingly the SYNGAP1 protein, stops neurons from sprouting too many connections. Without enough of this protein, young brains grow wild and unruly. Their brain circuits become extra sensitive to stimulus, impeding learning by failing to extract much useful information about the world. These overly connected brains are prone to epilepsy and a host of other symptoms.
Yet, how SYNGAP1 mutations affect specific circuits remains an active area of inquiry, says Gavin Rumbaugh, a neuroscientist at Scripps Research who once worked with Huganir. Rumbaugh’s research suggests the condition mutes rather than intensifies the response of sensory circuits such as touch. If that were true, it could possibly explain why Landen doesn't outwardly show distress. In fact, Rumbaugh speculates that intellectual disability may arise from a distortion of sensory information. “They may not experience the world in the same way as neurotypical children,” he says.
The story of SYNGAP1 began with Huganir and Caltech neuroscientist Mary Kennedy independently discovering the SYNGAP1 gene and protein in 1998 while trying to work out how the brain develops during infancy and how it processes information in adults. About a decade later, their work leapt from lab to clinic when a genetic analysis of three individuals linked their intellectual disability with mutations in their SYNGAP1 gene. At that moment, Rumbaugh decided to take his recently formed neuroscience lab in a more clinical direction. “That day I said, ‘let’s model this in mice and try to understand what happens to brain development when you lose one copy of SYNGAP1,’” he recalls.
![]()
From the patient side, increased accessibility to genetic sequencing gave families a name for their children’s disorders but little else. One parent, Monica Weldon, grew a Facebook group into Bridge the Gap, a foundation that keeps families updated on research and maintains a patient registry to connect SYNGAP1 children with researchers. The database, where parents log symptoms and other information, helps researchers connect dots that would otherwise be hard to see from the relatively small number of published case studies. Database reports of children like Landen not responding to pain, for instance, inspired Rumbaugh to investigate sensory circuits.
Recently Rumbaugh and Huganir have taken complimentary approaches to improving the lives of people with SYNGAP1 mutations. Rumbaugh seeks a drug that will restore normal levels of the SYNGAP1 protein. Such a treatment could help infants with SYNGAP1 mutations develop more normal neural wiring, and recent research in mice suggests that boosting the SYNGAP1 protein could also decrease seizure rates in older children and adults.
Huganir is looking at ways to curb the activity of a protein group called RAS. SYNGAP1 mutations activate RAS proteins. Inhibiting the activity of RAS proteins could stop neurons from getting so overdeveloped. Unpublished work and two case studies suggest that the statin drugs used to lower cholesterol may interfere with RAS activity and diminish the havoc caused by SYNGAP1 mutations. He is also looking at ways to restore normal SYNGAP protein expression.
Longer term, Huganir hopes gene editing tools will mature to the point where they can correct the genetic typo from the very beginning, sidestepping the need to fiddle with proteins in developing brains. “Those are approaches that lots of people are thinking about,” he says, “and probably will be viable in the next five to 10 years.”
About the Author

Charlie Wood is a science writer with a bachelor’s degree in physics from Brown University and a master’s degree in science journalism from New York University. In previous lives he taught physics in Mozambique and English in Japan, but these days he freelances from his home in New York.
CONTENT PROVIDED BY
BrainFacts/SfN
Discussion Questions
- How were the SYNGAP1 gene and protein discovered?
- What questions are neuroscientists still trying to answer about SYNGAP1?
- What innovations are neuroscientists hoping to develop in the future for SYNGAP1 treatment?
References
(2019, April 30) SYNGAP1-related intellectual disability. Retrieved from https://ghr.nlm.nih.gov/condition/syngap1-related-intellectual-disability
Clement, J. P., Aceti, M., Creson, T. K., Ozkan, E. D., Shi, Y., Reish, N. J., … Rumbaugh, G. (2012). Pathogenic SYNGAP1 Mutations Impair Cognitive Development by Disrupting Maturation of Dendritic Spine Synapses. Cell, 151(4), 709–723. doi: 10.1016/j.cell.2012.08.045
Cook, E. H., Masaki, J. T., Guter, S. J., & Najjar, F. (2019). Lovastatin Treatment of a Patient with a De Novo SYNGAP1 Protein Truncating Variant. Journal of Child and Adolescent Psychopharmacology, 29(4), 321–322. doi: 10.1089/cap.2018.0159
Hamdan, F. F., Gauthier, J., Spiegelman, D., Noreau, A., Yang, Y., Pellerin, S., … Michaud, J. L. (2009). Mutations in SYNGAP1 in Autosomal Nonsyndromic Mental Retardation. New England Journal of Medicine, 360(6), 599–605. doi: 10.1056/NEJMoa0805392
Michaelson, S. D., Ozkan, E. D., Aceti, M., Maity, S., Llamosas, N., Weldon, M., … Rumbaugh, G. (2018). SYNGAP1 heterozygosity disrupts sensory processing by reducing touch-related activity within somatosensory cortex circuits. Nature Neuroscience, 21(12), 1–13.
doi: 10.1038/s41593-018-0268-0
The Deciphering Developmental Disorders Study, Fitzgerald, T. W., Gerety, S. S., Jones, W. D., van Kogelenberg, M., King, D. A., … Hurles, M. E. (2014). Large-scale discovery of novel genetic causes of developmental disorders. Nature, 519, 223–228. doi: 10.1038/nature14135
Weldon, M., Kilinc, M., Lloyd Holder, J., & Rumbaugh, G. (2018). The first international conference on SYNGAP1-related brain disorders: a stakeholder meeting of families, researchers, clinicians, and regulators. Journal of Neurodevelopmental Disorders, 10(1), 6.
doi: 10.1186/s11689-018-9225-1