Grasping Alzheimer's Disease
- Reviewed10 Apr 2023
- Author Clinton Parks
- Source BrainFacts/SfN

Alzheimer’s disease is a form of dementia that is eventually fatal. Over time, this devastating disease causes a person’s brain to undergo irreversible, progressive degeneration, impairing their memory and reasoning. Despite its prevalence, researchers have much to learn about how the disease arises, how to treat it, and ultimately how to prevent it.
Alzheimer’s disease can be divided into less common early-onset forms and more typical late-onset forms. Patients with late-onset Alzheimer’s display symptoms in their mid-60s or later, with symptoms becoming more severe with age. In idiopathic early-onset forms of the disease, which account for around 10% of all cases, patients can start to experience symptoms in their 40s-50s. Autosomal dominant early-onset forms can begin in patients in as early as their 30s, although this makes up less than 1% of all Alzheimer’s cases.
Prevalence and Impact
Alzheimer’s is the most common cause of dementia in older adults. Of the 55 million people with dementia worldwide, approximately 60% to 70% have Alzheimer’s. The disease affects 5% to 8% of all people over 65 years of age, 15% to 20% above 75, and 25% to 50% of those over 85. It’s estimated that more than 6 million people in the U.S. suffer from the disease; however, the actual number could be as many as 11 million, including those who are asymptomatic. According to conservative estimates, Alzheimer’s will affect 13.8 million people in the U.S. by 2050.
In 2021, Alzheimer’s was the seventh leading cause of death in the U.S., accounting for 119,399 deaths. Deaths rose from 16.5 per 100,000 people in 1999 to 36 per 100,000 in 2021, but Alzheimer’s-related deaths are believed to be severely underreported. Some patients go undiagnosed, and others have dementia-related conditions (such as aspiration pneumonia) rather than Alzheimer’s listed as their primary cause of death. Some estimate that the number of Alzheimer’s-related deaths might be six to seven times higher than reported figures. If that is the case, it would be the third leading cause of death among older Americans.
Symptoms of Alzheimer's Disease
The stages of Alzheimer’s are classified by the symptoms patients experience. Early-stage symptoms include memory problems (more than expected in healthy people of a similar age), difficulty concentrating or finding appropriate words, and disorientation in time or place. Most people are not diagnosed until the mild stage when symptoms include personality and behavior changes, wandering and getting lost, repeating questions, losing and placing objects in odd places, taking longer to complete daily tasks, and having trouble handling money and paying bills. In the moderate stage, some patients have trouble recognizing family and friends; inability to learn new things; problems coping with new situations; difficulty getting dressed or performing other multistep tasks; hallucinations, delusions, and paranoia; and impulsive behavior. In the severe stage, patients are completely dependent on others for care, as their body begins shutting down. Their communication is reduced to groans, moans, and grunts; sleeping increases; and they become bedridden. Other severe-stage symptoms include weight loss, seizures, difficulty swallowing, skin infections, and a lack of bowel and bladder control.
Diagnosing Alzheimer’s
Alzheimer’s dementia is most commonly diagnosed by a physician asking the patient and a family member or friend about the patient’s health, medical history, ability to perform daily activities, and changes in behavior and personality. The physician then conducts tests on memory, problem-solving, attention, counting, and language. Even if mental deficits are found, including dementia, the condition still might not be Alzheimer’s. Similar deficits could be due to other conditions, including Lewy body disease, frontotemporal dementia, Parkinson’s disease, stroke, a tumor, sleep disturbances, side effects from medication, or infection.
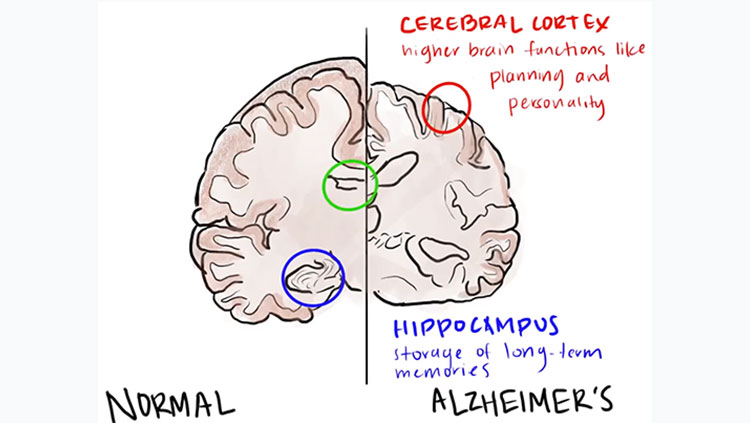
Ideally, physicians would be able to rely on a simple blood or urine test to diagnose Alzheimer’s. Research to identify a defining biomarker for Alzheimer’s — a specific biological indicator whose presence can identify a disease — is ongoing. Two candidate primary biomarkers are proteins called amyloid-beta (also called beta-amyloid) and tau. In Alzheimer’s, amyloid-beta forms into malformed clumps of protein called extracellular senile plaques or neuritic plaques. The disease also features abnormalities in a protein called tau, which normally stabilizes the cellular skeleton, where tau forms neurofibrillary tangles inside neurons. Both abnormalities are found in the brains of people with Alzheimer’s but, at present, no definitive biomarker can diagnose the disease in its early stages. A diagnosis can only be confirmed by postmortem examination.
Some potential diagnostic methods for Alzheimer’s include brain imaging, genetic risk profiling, and examining cerebrospinal fluid or blood. Neuroimaging, among the most promising areas of research focused on early detection, uses a mildly radioactive chemical marker that binds to amyloid plaques and shows their location in PET scans of living people. Starting in 2012, the FDA began approving use of molecular imaging tracers in combination with positron emission tomography (PET) to evaluate possible Alzheimer’s disease or other causes of dementia. To date, the FDA has approved the use of three tracers — florbetapir, flutemetamol, and florbetaben — which emit low levels of radioactivity that can be measured during PET scans to detect amyloid-beta and another tracer — flortaucipir — to detect tau protein clumps in the brain. However, neuritic plaques and neurofibrillary tangles are also present in the brains of people with no dementia or Alzheimer’s. So these scans are not used for routine evaluation, but are routinely used in clinical trials as surrogate markers for the disease. Studies to evaluate the benefit of such biomarkers in routine care are ongoing.
Causes and Pathology
The causes and mechanisms underlying Alzheimer’s disease are not fully understood. Most forms are likely caused by a combination of heredity, environment, and habits. Evidence has been building that repeated head trauma is one contributing factor increasing Alzheimer’s disease risk.
Patients experience the first cellular changes associated with Alzheimer’s a decade or more before becoming symptomatic. The neuronal transport system shows damage early in Alzheimer’s. Patients produce fewer neurotransmitters — chemical molecules that are released from an axon terminal, travel across gaps called synapses, and transmit signals to another neuron, organ, or other tissue type. In Alzheimer’s disease, axons and synapses are damaged and ultimately destroyed. Damage to neuronal transport impairs attention, memory, learning, and higher cognitive abilities. While the cause is unknown, neuritic plaques and neurofibrillary tangles are the two prime suspects. Plaques consist of amyloid-beta, which is formed from malformed clumps of a fragment of amyloid precursor protein (APP), a fibrous protein often found at neuronal synapses. In its soluble form, amyloid-beta can bind strongly to neural receptors, which initiates the erosion of synapses. Evidence indicates that this soluble form is highly toxic for synapses, while the insoluble form (which has low toxicity) tends to aggregate and is found in much higher concentrations than the soluble form. Some research suggests that the highly toxic, soluble form would be a better target for effective therapies.
Researchers are working to pinpoint the sequence of events that leads to Alzheimer’s. For many years, the amyloid hypothesis has been the dominant theory of how beta amyloid and tau protein interact to cause Alzheimer’s. This hypothesis asserts that amyloid-beta starts a sequence of events that ultimately lead to Alzheimer’s disease. Amyloid-beta accumulations first appear in the neocortex. Its neurotoxicity might be due to the fact that it exacerbates oxidative stress and damages the mitochondria, the cell’s primary energy supply unit, initiating a cascade of neuronal dysfunction and cell death. The formation of neuritic plaques induces tau proteins to become defective and tangle into neurotoxic neurofibrillary tangles (hyperphosphorylated tau protein) within neuron cell bodies. (In contrast, normal tau protein stabilizes microtubules, which are crucial to axonal transport.) Neurofibrillary tangles are generally first seen in the entorhinal cortex and the hippocampus, regions responsible for short-term memory and for transferring those memories to longer-term memory.
Although amyloid-beta and tau accumulations are found in people with Alzheimer’s, there is no definite proof that they cause Alzheimer’s. We do have evidence that tau and amyloid-beta might interact before clumping into their recognized disease forms. Even before it aggregates, malfunctioning tau can damage cellular transportation by blocking the microtubule tracks. Also, high tau levels can impair the function of amyloid-beta. These facts suggest a more central role for tau protein in the development of Alzheimer’s than generally recognized by the amyloid hypothesis.
It’s possible that inflammation, the presence of obesity, and cardiovascular disease can trigger these protein changes, increasing Alzheimer’s incidence and severity. Plaques and tangles are known to negatively interact with microglia, non-neural brain cells that act as immune cells for the central nervous system, and astroglia, which offer physiological regulation and structural support in the brain.
Genetics of Alzheimer’s
Early-onset Alzheimer’s is a rare, dominantly inherited form of the disease. This means that a defect in only one of the two copies of a gene housed in our chromosomes results in disease. Three different genes — APP, PSEN1, and PSEN2 — can cause early-onset familial Alzheimer’s disease, which starts when people are in their 40s and 50s, through dominant inheritance. In late-onset Alzheimer’s, the ApoE4 variant of the Apolipoprotein E (APOE) gene is a major genetic risk factor but not a determining one. The normal protein, ApoE, is mainly produced by astroglia or damaged neurons and helps clear soluble amyloid-beta from the brain.
In most people, Alzheimer’s results from a combination of genetic and environmental causes. Several genetic associations have been noted. A mutant C9ORF72 gene has been found in people with both early- and late-onset forms of the disease. This gene codes for a protein that regulates transportation in the intracellular matrix. The mutation was already known to play a major role in ALS and frontotemporal dementia.
The TOMM40 gene, which codes for a protein responsible for moving proteins into mitochondria, has a complex relationship with Alzheimer’s. People with a longer version of the gene were shown to be either predisposed or resistant to Alzheimer’s — depending on whether a parent had the disease. Among those with an afflicted parent, people with the longer version of the gene were more apt to develop dementia than those with the shorter allele; but among those with no afflicted parent, people with a longer version of the gene displayed better memory than those with a shorter version.
With the TREM2 gene, loss-of-function mutations cause a sequence of physiological events associated with Alzheimer’s disease. This suggests a possible genetic link between early- and adult-onset variants. Early-onset Alzheimer’s is linked to patients who have mutations that cause loss of function of both copies of the TREM2 gene, whereas adult-onset disease is associated with the loss of function in one copy of the gene. Normally, TREM-2 protein helps regulate removal of cell debris, clearing amyloid proteins, and suppressing inflammation in microglia.
Treatments for Alzheimer’s
The U.S. FDA has now approved seven prescription drugs for treating Alzheimer’s. While they relieve some symptoms, or possibly slow disease progression, none cure the disease. Three of these drugs are cholinesterase inhibitors: donepezil, galantamine, and rivastigmine. Cholinesterase inhibitors stop the action of acetylcholinesterase, an enzyme that breaks down the neurotransmitter acetylcholine. This increases the available amount of acetylcholine (involved in learning and memory), which counteracts the damaging effect of the disease on production of this neurotransmitter.
The fourth drug, memantine, is an NMDA receptor antagonist. Normally, NMDA receptors bind the neurotransmitter glutamate, allowing calcium to enter the neuron. In Alzheimer’s, the damaged cells become overwhelmed with calcium, further damaging the neurons — a condition called neuronal excitotoxicity. Memantine blocks the flow of calcium through NMDA-receptor channels.
The fifth approved medication combines donepezil and memantine. Donepezil can be used in all stages of the disease, galantamine for mild to moderate stages, memantine for moderate to severe stages, and rivastigmine in all stages. The donepezil/memantine cocktail is used to treat moderate to severe Alzheimer’s.
The sixth and seventh approved medications, aducanumab and lecanemab, are both monoclonal antibodies which target amyloid-beta. Monoclonal antibodies are lab-derived proteins which act like cells in our immune system, seeking out specific materials called antigens to stick to them and destroy them. “Monoclonal” refers to the antibodies being exact copies of one lab-derived antibody. Both drugs were granted accelerated approval by the FDA — aducanumab in June 2021, and lecanemab in January 2023. While aducanumab reduced the amount of amyloid-beta in the brain, it had no effect on cognition or the progress of the disease. Lecanemab resulted in slowing the progression of the disease by 27%. Patient use of these drugs and further study will shed light on their effectiveness for treating Alzheimer’s disease.
Adapted from the 8th edition of Brain Facts by Clinton Parks.
About the Author

Clinton Parks is a Washington, DC-based freelance science writer who writes for AAAS’ ScienceCareers, American Chemical Society’s Axial, and American Physical Society’s Physics Buzz, among others.
CONTENT PROVIDED BY
BrainFacts/SfN
References
Alzheimer’s Disease Fact Sheet. (2017). National Institute on Aging. NIH. https://www.nia.nih.gov/health/alzheimers-disease-fact-sheet
Bischof, G. N., Bartenstein, P., Barthel, H., van Berckel, B., Doré, V., van Eimeren, T., Foster, N., Hammes, J., Lammertsma, A. A., Minoshima, S., Rowe, C., Sabri, O., Seibyl, J., Van Laere, K., Vandenberghe, R., Villemagne, V., Yakushev, I., & Drzezga, A. (2021). Toward a Universal Readout for 18F-Labeled Amyloid Tracers: The CAPTAINs Study. Journal of Nuclear Medicine. 62(7), 999–1005. https://doi.org/10.2967/jnumed.120.250290
Borel, F., Gernoux, G., Cardozo, B., Metterville, J. P., Toro Cabrera, G. C., Song, L., Su, Q., Gao, G. P., Elmallah, M. K., Brown, R. H., Jr, & Mueller, C. (2016). Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1(G93A) Mice and Nonhuman Primates. Human gene therapy, 27(1), 19–31. https://doi.org/10.1089/hum.2015.122
Chronic Traumatic Encephalopathy (CTE). (n.d.) Related Conditions. Alzheimer’s Association. https://www.alz.org/alzheimers-dementia/what-is-dementia/related_conditions/chronic-traumatic-encephalopathy-(cte)
Cummings, J. L., Morstorf, T. & Zhong, K. (2014). Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alz Res Therapy 6, 37. https://doi.org/10.1186/alzrt269
de Oliveira, G., Marques, M., Cruzeiro-Silva, C. et al. Structural basis for the dissociation of α-synuclein fibrils triggered by pressure perturbation of the hydrophobic core. Sci Rep 6, 37990 (2016). https://doi.org/10.1038/srep37990
Dementia. (2017). Fact Sheets. World Health Organization. https://www.who.int/en/news-room/fact-sheets/detail/dementia
Dementia statistics. (2023). Numbers of people with dementia. Alzheimer’s Disease International. https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/
DiSalvo, D. (June 2017). Alzheimer's Affects Twice As Many People As Estimated, And The Numbers Are Climbing. Forbes. https://www.forbes.com/sites/daviddisalvo/2017/06/15/alzheimers-affects-twice-as-many-people-as-estimated-suggests-new-study/?sh=2e7add3d644a
Dorsey, E. R., Constantinescu, R., Thompson, J. P., Biglan, K. M., Holloway, R. G., Kieburtz, K., Marshall, F. J., Ravina, B. M., Schifitto, G., Siderowf, A., Tanner, C. M. (Jan 2007). Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology, 68 (5) 384-386; DOI: 10.1212/01.wnl.0000247740.47667.03
Earlier Diagnosis. (n.d.) Research and Progress. Alzheimer’s Association. https://www.alz.org/alzheimers-dementia/research_progress/earlier-diagnosis
Family history of Alzheimer’s may alter metabolic gene that increases risk for disease. (May 2017). Iowa State University of Science and Technology. https://www.news.iastate.edu/news/2017/05/22/alzheimersgene
FDA Approves First Drug to Image Tau Pathology in Patients Being Evaluated for Alzheimer’s Disease. (2020). FDA. https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-image-tau-pathology-patients-being-evaluated-alzheimers-disease
Fernández, A., Llacuna, L., Fernández-Checa, J. C., & Colell, A. (2009). Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. The Journal of neuroscience: the official journal of the Society for Neuroscience, 29(20), 6394–6405. https://doi.org/10.1523/JNEUROSCI.4909-08.2009
Gale, C., & Martyn, C. (2003). Tobacco, coffee, and Parkinson's disease. BMJ (Clinical research ed.), 326(7389), 561–562. https://doi.org/10.1136/bmj.326.7389.561
Grainger, D. (January 2015). Why Too Many Clinical Trials Fail -- And A Simple Solution That Could Increase Returns On Pharma R&D. Forbes. https://www.forbes.com/sites/davidgrainger/2015/01/29/why-too-many-clinical-trials-fail-and-a-simple-solution-that-could-increase-returns-on-pharma-rd/?sh=a8ba6bedb8b3
Goldman, B. (September 2013). Scientists reveal how beta-amyloid may cause Alzheimer's. Stanford Medicine. https://med.stanford.edu/news/all-news/2013/09/scientists-reveal-how-beta-amyloid-may-cause-alzheimers.html
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., Cruchaga, C., Sassi, C., Kauwe, J. S., Younkin, S., Hazrati, L., Collinge, J., Pocock, J., Lashley, T., Williams, J., Lambert, J. C., Amouyel, P., Goate, A., Rademakers, R., Morgan, K., … (2013). Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer's disease. The New England Journal of Medicine, 368(2), 117–127. https://doi.org/10.1056/NEJMoa1211851
He, W., Goodkind, D., Kowal, P. (March 2016). An Aging World: 2015. 5th Edition. United States Census Bureau. https://web.archive.org/web/20230404232414/https://www.census.gov/newsroom/press-releases/2016/cb16-54.html
Hebert, L. E., Weuve, J., Scherr, P. A., Evans, D. A. (May 2013). Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology, 80 (19) 1778-1783; DOI:10.1212/WNL.0b013e31828726f5
Henchcliffe, C., & Beal, M. F. (2008). Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nature clinical practice. Neurology, 4(11), 600–609. https://doi.org/10.1038/ncpneuro0924
Heneka, M. T., Carson, M. J., Khoury, J. E., Landreth, G. E., Brosseron, F., et. al. (2015) Neuroinflammation in Alzheimer’s Disease. The Lancet Neurology. 14 (4) 388-405. https://doi.org/10.1016/S1474-4422(15)70016-5.
Jonas E. A. (2014). Impaired import: how huntingtin harms. Nature Neuroscience, 17(6), 747–749. https://doi.org/10.1038/nn.3726
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., Burrell, J. R., & Zoing, M. C. (2011). Amyotrophic lateral sclerosis. Lancet (London, England), 377(9769), 942–955. https://doi.org/10.1016/S0140-6736(10)61156-7
Kikuchi, T., Morizane, A., Doi, D. et al. (2017). Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature, 548, 592–596. https://doi.org/10.1038/nature23664
Kochanek, K. D., Murphy, S. L., Xu, J., and Tejada-Vera, B. (June 2016). Deaths: Final Data for 2014. National Vital Statistics Reports, 65 (4) 1-122. Centers for Disease Control. https://www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_04.pdf
Lees, A. J., Hardy, J., & Revesz, T. (2009). Parkinson's disease. Lancet (London, England), 373(9680), 2055–2066. https://doi.org/10.1016/S0140-6736(09)60492-X
Lemere, C. A. Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol Neurodegeneration, 8, 36 (2013). https://doi.org/10.1186/1750-1326-8-36
Lue, L. F., Kuo, Y. M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., Beach, T., Kurth, J. H., Rydel, R. E., & Rogers, J. (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. The American journal of pathology, 155(3), 853–862. https://doi.org/10.1016/s0002-9440(10)65184-x
McGinley, L. (January 2023). Alzheimer’s drug that slows cognitive decline gets FDA approval. The Washington Post. https://www.washingtonpost.com/health/2023/01/06/alzheimers-drug-lecanemab-fda-approval/
Monoclonal Antibodies. (February 2023). Cleveland Clinic. https://my.clevelandclinic.org/health/treatments/22246-monoclonal-antibodies
Niemelä, V., Landtblom, A. M., Blennow, K., & Sundblom, J. (2017). Tau or neurofilament light-Which is the more suitable biomarker for Huntington's disease?. PloS one, 12(2), e0172762. https://doi.org/10.1371/journal.pone.0172762
Number of Alzheimer's deaths found to be underreported. (May 2014). National Institute on Aging. NIH. https://www.nia.nih.gov/news/number-alzheimers-deaths-found-be-underreported
Padda IS, Parmar M. Aducanumab. (November 2022). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK573062/
Parkinson’s Disease. (n.d.) Harvard Stem Cell Institute. The President and Fellows of Harvard College. https://hsci.harvard.edu/parkinsons-disease-0
Parkinson's Disease: Hope Through Research. (n.d) National Institute of Neurological Disorders and Stroke. NIH. https://www.ninds.nih.gov/health-information/patient-caregiver-education/hope-through-research/parkinsons-disease-hope-through-research
Phase 3 Clinical Trial Targeting Lou Gehrig's Disease Gets $15.9 Million Investment From Stem Cell Agency. (July 2017). California Insititute for Regenerative Medicine. https://www.cirm.ca.gov/about-cirm/newsroom/press-releases/07202017/phase-3-clinical-trial-targeting-lou-gehrigs-disease
Reardon, S. (2023). FDA approves Alzheimer’s drug lecanemab amid safety concerns. Nature. https://www.nature.com/articles/d41586-023-00030-3
Schulte, J., & Littleton, J. T. (2011). The biological function of the Huntingtin protein and its relevance to Huntington's Disease pathology. Current Trends in Neurology, 5, 65–78. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3237673/
Shugart, J. (July 2017). C9ORF72 Throws a Wrench into DNA Repair Machinery. Alzforum. FBRI, LLC. https://www.alzforum.org/news/research-news/c9orf72-throws-wrench-dna-repair-machinery
Sun, X., Chen, W. D., & Wang, Y. D. (2015). β-Amyloid: the key peptide in the pathogenesis of Alzheimer's disease. Frontiers in Pharmacology, 6, 221. https://doi.org/10.3389/fphar.2015.00221
The Ionis antisense pipeline. (n.d.) Ionis Pharmaceuticals, Inc. https://www.ionispharma.com/ionis-innovation/pipeline/
van Dyck, C. H., Swanson, C. J., Aisen, P., Bateman, R. J., Chen, C., Gee, M., Kanekiyo, M., Li, D., Reyderman, L., Cohen, S., Froelich, L., Katayama, S., Sabbagh, M., Vellas, B., Watson, D., Dhadda, S., Irizarry, M., Kramer, L. D., & Iwatsubo, T. (2023). Lecanemab in Early Alzheimer's Disease. The New England journal of medicine, 388(1), 9–21. https://doi.org/10.1056/NEJMoa2212948
van Rheenen, W., Shatunov, A., Dekker, A. et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48, 1043–1048. https://doi.org/10.1038/ng.3622
Xu, J. (September 2015). QuickStats: Age-Adjusted Death Rates for Parkinson Disease — United States, 2000–2013. Morbidity and Mortality Weekly Report. National Vital Statistics System, 64 (36);1034. Mortality public use data files, 2000–2013. Centers for Disease Control and Prevention. https://www.cdc.gov/mmWr/preview/mmwrhtml/mm6436a9.htm
What are the Signs of Alzheimer’s Disease? (2017). National Institute on Aging. NIH. https://www.nia.nih.gov/health/what-are-signs-alzheimers-disease
What is ALS? (n.d.) The ALS Association. https://www.als.org/understanding-als/what-is-als

.jpg)