Pain Therapies
- Published25 Oct 2016
- Reviewed25 Oct 2016
- Author Stephani Sutherland
- Source BrainFacts/SfN
Pain serves a vital purpose, drawing our attention to tissue damage to prevent further damage and help us heal after injury. But for some pain becomes a chronic condition, and opioids — the most potent pain relievers available — can cause devastating side effects.
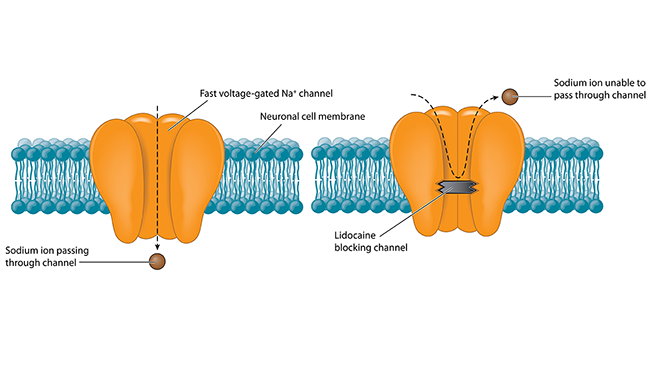
The flux of charged atoms in and out of neurons drives electrical signaling. Sodium ions pass through tiny pores in the cell membrane called sodium channels. Anesthetic drugs like lidocaine squelch pain signals at the source by blocking these channels.
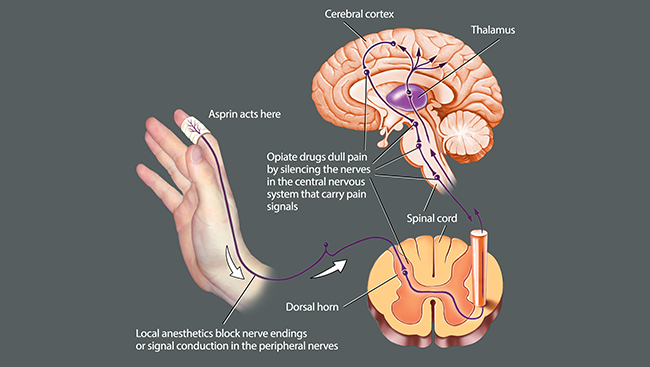
At the site of an injury, the body produces compounds called prostaglandins, which increase pain sensitivity. Aspirin relieves pain by halting the production of prostaglandins. Other analgesics work in the nervous system to relieve pain: Local anesthetics intercept pain signals traveling up the nerve and opiate drugs block the transfer of pain signals from the spinal cord to the brain.
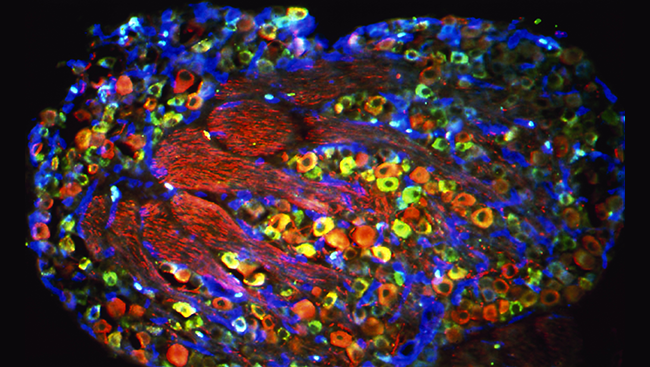
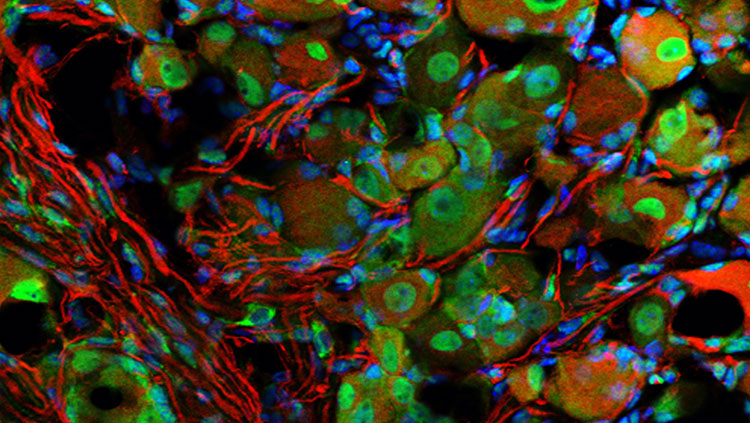
Located throughout the body, pain-sensing neurons transmit pain signals to the spinal cord. Their cell bodies — and the cell bodies of other sensory neurons — cluster in structures alongside the spinal cord called dorsal root ganglia. This cross section of one dorsal root ganglion shows the cell bodies of touch-sensing neurons (red) and pain-sensing neurons (green and blue).
Efforts to alleviate pain are as old as humanity. Opioids, the most potent pain relievers, have been used for millennia despite associated risks for addiction and death. New efforts to target pain at its source are offering hope that we can alleviate pain without devastating side effects.
A Channel for Pain
Pain is an essential part of being human. And, attempts to relieve pain date back to prehistory. For millennia humans have regarded opium and its derivatives as the most effective drug to alleviate pain: archaeologists unearthed evidence that Neolithic farmers cultivated opium poppies near Rome 8,000 years ago, and poppy cultivation has been documented in ancient Asia, Mesopotamia, Greece, and Egypt. Throughout history, using opium and its modern derivatives has been a double-edged sword, relieving pain while risking a dangerous addiction.
Pain begins with specialized sensory nerves that reach throughout the body detecting damage. Like all neurons, these cells send messages via electrical signals that develop from the flux of charged atoms in and out of the cell. Voltage-gated ion channels, tiny proteins embedded in the cell membrane, control the flow of specific ions like sodium and potassium in response to voltage changes. Channels marshaling sodium ions into cells are called NaVs — Na the chemical shorthand for sodium and V for voltage. Blocking NaVs stops the flow of sodium and, among other things, squelches pain signals, as anyone receiving lidocaine from a dentist can attest. But different types of NaVs act throughout the brain and heart; blocking every NaV would shut down the body’s vital functions. The only way to dampen pain safely is to target whatever NaV exists only in sensory neurons.
By the 1990s, researchers isolated the sodium channels responsible for electrical signaling in heart, muscle and brain, but they still had not found the predominant NaV in sensory nerves. That changed in 1997, when researchers led by Gail Mandel at State University of New York at Stony Brook identified a sodium channel they called peripheral nerve channel 1 (PN1) concentrated at the ends of rat sensory neurons where pain signals start. Today, PN1 — now known as NaV1.7 — is at the center of intense efforts to develop new pain drugs. Once NaV1.7 had been identified as a primary generator of sensory signals, the hunt was on for agents that could block or reduce the channel’s activity in hopes of dampening pain.
Learning From Inherited Pain Syndromes
Imagine a life completely free of pain. No toothaches. No searing muscle aches. No gasps from grabbing a hot pan. Such is life for people with congenital insensitivity to pain (CIP), an exceedingly rare condition first seen in families in Pakistan. It may sound enticing, but pain serves a vital purpose: to minimize damage and to help us heal after injury. Without the protective sensation of pain, people with CIP can’t feel when they’ve been injured and can’t tell when that injury is serious. For example, children with CIP have chewed off parts of their tongues when they were teething because they couldn’t feel pain. As a result, people with CIP are at risk for serious injury and even death. Understanding their plight, however, has expanded our knowledge about pain.
By the early 2000’s, accumulating evidence from animal research and studies in cells linked NaV1.7 to pain transmission. In 2006, Geoff Woods and James Cox working at the University of Cambridge strengthened that link by demonstrating that members of a family with CIP carried a mutation in SCN9A making NaV1.7 nonfunctional. Without NaV1.7 activity, those patients lack the sensation of pain. The finding cemented a critical role for NaV1.7 in pain although exactly how NaV1.7 contributes to the perception of pain remains unknown.
Conversely, bolstering the link between NaV1.7 and pain, studies of people with rare pain disorders revealed that they too carried mutations in SCN9A — ones making NaV1.7 overly active, causing extreme pain. These disorders include a disease called erythromelalgia (EM), from the Greek for “red limb.” With EM, warmth or mild exercise can trigger an excruciating burning pain that turns the hands or feet red and swollen.
Glitches in the code for NaV1.7 do not just cause rare diseases. A 2012 collaboration between researchers at Yale University School of Medicine and University Medical Center in Maastricht, Netherlands, discovered some people with a common pain disorder broadly labeled small-fiber neuropathy (SFN) also harbored variations in SCN9A. Most importantly, the findings have deepened our understanding of pain and solidified the rationale for aiming drugs at NaV1.7.
New Avenues for Treating Pain
The world’s oldest and best-known pain-fighting drug is opium, derived from the poppy flower. Synthetic versions, called opioids, remain the gold standard of analgesics. For patients with the acute pain that comes after a severe injury or a surgery, nothing cuts pain like opioids. But for some people pain becomes a chronic agony, lingering for years. Physicians and scientists disagree about whether opioids relieve chronic pain, such as back pain, over the long term — and some evidence hints they make pain worse. What’s more, opioids cause side effects including constipation, addiction, and respiratory depression, which can kill. Unfortunately, over the past twenty years opioid use has spiked causing an epidemic of opioid drug misuse. Never has the need for new pain drugs been greater.
Opioids work by mimicking a suite of naturally occurring pain-relieving molecules produced by the brain. Like those brain molecules, opioids quiet pain signals in the brain and spinal cord, but not at its source — peripheral sensory neurons. Medications dampening NaV1.7, in contrast, would.
An early clinical trial of a NaV1.7 blocker in five people with inherited EM — a disease caused by SCN9A mutations — showed the drug dampened pain in these patients. While that drug blocked NaV1.7, it also interfered with other NaVs. As a result, it caused unacceptable side effects at the doses required to relieve more common types of pain. Elements of the pain transmission system possess high levels of NaV1.7 so it remains a promising drug target for new pain therapeutics, and pharmaceutical companies continue to develop more selective blockers. As clinical trials of one of these more selective NaV1.7 blockers has shown efficacy in treating trigeminal neuralgia, one of the most painful medical conditions known, NaV1.7 blockers might be available for general use within the next few years.
Importantly, blocking NaV1.7 activity could improve pain treatment either alone or in combination with other medications. That approach holds the promise of offering pain relief while reducing the use and misuse of opioids. Through continued research efforts funded by public and private institutions, scientists may soon reach this worthy goal.
About the Author

Stephani Sutherland is a neuroscientist and freelance journalist in southern California. She is a frequent contributor at Pain Research Forum and Scientific American.
CONTENT PROVIDED BY
BrainFacts/SfN
References
Cox JJ, Sheynin J, Shorer Z, Reimann F, Nicholas AK. Congenital insensitivity to pain: novel SCN9A missense and in-frame deletion mutations. Human Mutation. 31(9):E1670-86 (2010).
Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nature Reviews Neuroscience. 14(1):49-62 (2013).
Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, et al. Gain of function Naν1.7 mutations in idiopathic small fiber neuropathy. Annals of Neurology. 71(1):26-39 (2012).
McDonnell A, Schulman B, Ali Z, Dib-Hajj SD, Brock F, et al. Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain. 139(Pt 4):1052-65 (2016).
Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. The Institute of Medicine of the National Academies. National Academies Press. 2011.
Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, et al. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proceedings of the National Academies of Science of the United States of America. 94(4):1527-32.
Volkow ND. America’s addiction to opioids: Heroin and prescription drug abuse. Testimony to Congress. May 14, 2014.