Illuminating Huntington’s Disease
- Reviewed20 Apr 2023
- Author Clinton Parks
- Source BrainFacts/SfN
Huntington’s disease (HD) is a heritable disease that impairs voluntary movement and cognition. The disease afflicts three to twelve of 100,000 people of Caucasian descent but is less common among East Asian and African populations. The HD variant of the HTT gene is dominant; if one parent has a single copy of the HD gene variant and the other parent has normal HTT genes, a child has a 50 percent chance of inheriting the HD variant and developing the disease.
The most common form of HD begins earlier than most progressive brain diseases, becoming active when people are in their 30s and 40s. Death occurs 15 to 20 years after a patient becomes symptomatic. Juvenile HD begins in childhood or adolescence, occurring before the age of 20. Juvenile HD patients usually die 10 to 15 years after their symptoms appear.
Symptoms
Signs of HD begin with irritability, mood swings, depression, small involuntary movements (called chorea), poor coordination, and difficulty making decisions and learning new information. As the disease progresses, the chorea becomes more pronounced and patients have increasing trouble with voluntary movements like walking, speaking, and even swallowing. Their cognitive problems also worsen.
Around 90% of HD is adult onset. While juvenile HD onset displays the same symptoms as the adult form, it also includes slow movements, clumsiness, frequent falling, rigidity, slurred speech, drooling, and seizures. School performance declines as thinking and reasoning abilities become impaired, and seizures occur in 30 to 50% of children with this condition.
Clinical onset of HD is defined by motor symptoms because the psychiatric symptoms are heterogeneous and have high prevalence in the general population, so they are more difficult to link specifically with HD. However, mouse models of HD also display these changes, so we know they are really part of the disease rather than co-morbidities or a consequence of the stresses of being from an HD family. These symptoms can occur decades prior to motor onset.
Causes and Genetics
In 1993, Huntington’s disease was found to be caused by mutations in the HTT gene, which codes for the huntingtin protein located on chromosome 4. The protein likely interacts with other proteins involved in signaling, transport, binding to proteins and other structures, and protecting the cell from self-destruction.
The disease mutation involves an abnormal number of repeats of a three-part (trinucleotide or triplet) snippet of DNA in the HTT gene. This sequence of the nucleotides cytosine, adenine, and guanine (CAG) normally repeats 10 to 26 times but occurs more times in the mutation. Individuals with 27-35 repeats will not develop the disease but have a risk of passing it to their children through inter-generational expansion, and people with 36-39 repeats may or may not get HD and can have children with mutations that lead to disease onset. Repeats of 36-39 are found in around one in 400 people in the general population, and about 10% of HD cases have no family history. The disease continues across generations from both non-disease-causing and disease-causing sequence lengths. Individuals with 40 or more repeats will develop HD and have a 50% chance of passing it to children. In individuals with greater than 60 repeats, disease onset is typically before the age of 20 (juvenile onset HD).
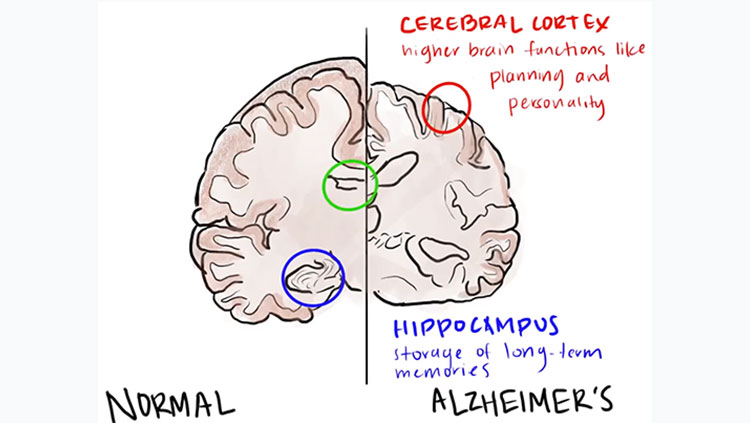
This expanded huntingtin protein is susceptible to clumping and can disrupt functions associated with normal huntingtin. However, some research suggests the aggregation of huntingtin proteins may provide protective mechanisms for neurons, with oligomers or other intermediate species potentially driving neural toxicity. Brain areas most often affected are the basal ganglia (voluntary movement) and the cortex (cognition, perception, and memory).
Treatments
There currently is no cure for Huntington’s disease, but the U.S. FDA has approved two treatments for associated symptoms.
In August of 2008, tetrabenazine was the first drug approved for treating chorea — involuntary jerking or writhing movements — associated with Huntington’s disease. Tetrabenazine is a monoamine-depleting agent for oral administration. Monoamine depletors generally work by reversibly exhausting one or more monoamine neurotransmitters like serotonin, dopamine, adrenaline, and noradrenaline.
In April 2017, the FDA approved another similar monoamine-depleting agent, deutetrabenazine, also for treating chorea associated with Huntington’s. Other emerging therapies are aimed at slowing the progression of Huntington’s disease by targeting the mutated form of the huntingtin protein produced in HD patients.
Furthermore, there is now evidence for viable HD biomarkers. Studies have suggested that elevated levels of neurofilament light chain (a component of the neuronal cytoskeleton), and to a lesser extent tau (which turns up regularly in neurodegenerative diseases), in cerebrospinal fluid are biomarkers of HD. Interestingly, neurofilament light chain is also being investigated as a biomarker for ALS and other neurodegenerative diseases. In mouse studies, the amount of neurofilament light chain found in the animals’ cerebral spinal fluid and blood increased before neurological signs appeared, likely coinciding with the development of brain lesions.
Other treatments for Huntington’s focus on ameliorating symptoms and addressing quality of life. For example, physicians treat chorea with antipsychotic and other medications; psychiatric disorders are addressed with antidepressents, antipsychotics, and mood-stabilizing drugs; psychotherapy, speech therapy, physical therapy, occupational therapy, lifestyle changes, and generalized care and support can improve the quality of life for people with Huntington’s disease.
Adapted from the 8th edition of Brain Facts by Clinton Parks.
About the Author

Clinton Parks is a Washington, DC-based freelance science writer who writes for AAAS’ ScienceCareers, American Chemical Society’s Axial, and American Physical Society’s Physics Buzz, among others.
CONTENT PROVIDED BY
BrainFacts/SfN
References
Alzheimer’s Disease Fact Sheet. (2017). National Institute on Aging. NIH. https://www.nia.nih.gov/health/alzheimers-disease-fact-sheet
Bischof, G. N., Bartenstein, P., Barthel, H., van Berckel, B., Doré, V., van Eimeren, T., Foster, N., Hammes, J., Lammertsma, A. A., Minoshima, S., Rowe, C., Sabri, O., Seibyl, J., Van Laere, K., Vandenberghe, R., Villemagne, V., Yakushev, I., & Drzezga, A. (2021). Toward a Universal Readout for 18F-Labeled Amyloid Tracers: The CAPTAINs Study. Journal of Nuclear Medicine. 62(7), 999–1005. https://doi.org/10.2967/jnumed.120.250290
Borel, F., Gernoux, G., Cardozo, B., Metterville, J. P., Toro Cabrera, G. C., Song, L., Su, Q., Gao, G. P., Elmallah, M. K., Brown, R. H., Jr, & Mueller, C. (2016). Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1(G93A) Mice and Nonhuman Primates. Human gene therapy, 27(1), 19–31. https://doi.org/10.1089/hum.2015.122
Chronic Traumatic Encephalopathy (CTE). (n.d.) Related Conditions. Alzheimer’s Association. https://www.alz.org/alzheimers-dementia/what-is-dementia/related_conditions/chronic-traumatic-encephalopathy-(cte)
Cummings, J. L., Morstorf, T. & Zhong, K. (2014). Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alz Res Therapy 6, 37. https://doi.org/10.1186/alzrt269
de Oliveira, G., Marques, M., Cruzeiro-Silva, C. et al. Structural basis for the dissociation of α-synuclein fibrils triggered by pressure perturbation of the hydrophobic core. Sci Rep 6, 37990 (2016). https://doi.org/10.1038/srep37990
Dementia. (2017). Fact Sheets. World Health Organization. https://www.who.int/en/news-room/fact-sheets/detail/dementia
Dementia statistics. (2023). Numbers of people with dementia. Alzheimer’s Disease International. https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/
DiSalvo, D. (June 2017). Alzheimer's Affects Twice As Many People As Estimated, And The Numbers Are Climbing. Forbes. https://www.forbes.com/sites/daviddisalvo/2017/06/15/alzheimers-affects-twice-as-many-people-as-estimated-suggests-new-study/?sh=2e7add3d644a
Dorsey, E. R., Constantinescu, R., Thompson, J. P., Biglan, K. M., Holloway, R. G., Kieburtz, K., Marshall, F. J., Ravina, B. M., Schifitto, G., Siderowf, A., Tanner, C. M. (Jan 2007). Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology, 68 (5) 384-386; DOI: 10.1212/01.wnl.0000247740.47667.03
Earlier Diagnosis. (n.d.) Research and Progress. Alzheimer’s Association. https://www.alz.org/alzheimers-dementia/research_progress/earlier-diagnosis
Family history of Alzheimer’s may alter metabolic gene that increases risk for disease. (May 2017). Iowa State University of Science and Technology. https://www.news.iastate.edu/news/2017/05/22/alzheimersgene
FDA Approves First Drug to Image Tau Pathology in Patients Being Evaluated for Alzheimer’s Disease. (2020). FDA. https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-image-tau-pathology-patients-being-evaluated-alzheimers-disease
Fernández, A., Llacuna, L., Fernández-Checa, J. C., & Colell, A. (2009). Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. The Journal of neuroscience: the official journal of the Society for Neuroscience, 29(20), 6394–6405. https://doi.org/10.1523/JNEUROSCI.4909-08.2009
Gale, C., & Martyn, C. (2003). Tobacco, coffee, and Parkinson's disease. BMJ (Clinical research ed.), 326(7389), 561–562. https://doi.org/10.1136/bmj.326.7389.561
Grainger, D. (January 2015). Why Too Many Clinical Trials Fail -- And A Simple Solution That Could Increase Returns On Pharma R&D. Forbes. https://www.forbes.com/sites/davidgrainger/2015/01/29/why-too-many-clinical-trials-fail-and-a-simple-solution-that-could-increase-returns-on-pharma-rd/?sh=a8ba6bedb8b3
Goldman, B. (September 2013). Scientists reveal how beta-amyloid may cause Alzheimer's. Stanford Medicine. https://med.stanford.edu/news/all-news/2013/09/scientists-reveal-how-beta-amyloid-may-cause-alzheimers.html
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., Cruchaga, C., Sassi, C., Kauwe, J. S., Younkin, S., Hazrati, L., Collinge, J., Pocock, J., Lashley, T., Williams, J., Lambert, J. C., Amouyel, P., Goate, A., Rademakers, R., Morgan, K., … (2013). Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer's disease. The New England Journal of Medicine, 368(2), 117–127. https://doi.org/10.1056/NEJMoa1211851
He, W., Goodkind, D., Kowal, P. (March 2016). An Aging World: 2015. 5th Edition. United States Census Bureau. https://web.archive.org/web/20230404232414/https://www.census.gov/newsroom/press-releases/2016/cb16-54.html
Hebert, L. E., Weuve, J., Scherr, P. A., Evans, D. A. (May 2013). Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology, 80 (19) 1778-1783; DOI:10.1212/WNL.0b013e31828726f5
Henchcliffe, C., & Beal, M. F. (2008). Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nature clinical practice. Neurology, 4(11), 600–609. https://doi.org/10.1038/ncpneuro0924
Heneka, M. T., Carson, M. J., Khoury, J. E., Landreth, G. E., Brosseron, F., et. al. (2015) Neuroinflammation in Alzheimer’s Disease. The Lancet Neurology. 14 (4) 388-405. https://doi.org/10.1016/S1474-4422(15)70016-5.
Jonas E. A. (2014). Impaired import: how huntingtin harms. Nature Neuroscience, 17(6), 747–749. https://doi.org/10.1038/nn.3726
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., Burrell, J. R., & Zoing, M. C. (2011). Amyotrophic lateral sclerosis. Lancet (London, England), 377(9769), 942–955. https://doi.org/10.1016/S0140-6736(10)61156-7
Kikuchi, T., Morizane, A., Doi, D. et al. (2017). Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature, 548, 592–596. https://doi.org/10.1038/nature23664
Kochanek, K. D., Murphy, S. L., Xu, J., and Tejada-Vera, B. (June 2016). Deaths: Final Data for 2014. National Vital Statistics Reports, 65 (4) 1-122. Centers for Disease Control. https://www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_04.pdf
Lees, A. J., Hardy, J., & Revesz, T. (2009). Parkinson's disease. Lancet (London, England), 373(9680), 2055–2066. https://doi.org/10.1016/S0140-6736(09)60492-X
Lemere, C. A. Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol Neurodegeneration, 8, 36 (2013). https://doi.org/10.1186/1750-1326-8-36
Lue, L. F., Kuo, Y. M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., Beach, T., Kurth, J. H., Rydel, R. E., & Rogers, J. (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. The American journal of pathology, 155(3), 853–862. https://doi.org/10.1016/s0002-9440(10)65184-x
McGinley, L. (January 2023). Alzheimer’s drug that slows cognitive decline gets FDA approval. The Washington Post. https://www.washingtonpost.com/health/2023/01/06/alzheimers-drug-lecanemab-fda-approval/
Monoclonal Antibodies. (February 2023). Cleveland Clinic. https://my.clevelandclinic.org/health/treatments/22246-monoclonal-antibodies
Niemelä, V., Landtblom, A. M., Blennow, K., & Sundblom, J. (2017). Tau or neurofilament light-Which is the more suitable biomarker for Huntington's disease?. PloS one, 12(2), e0172762. https://doi.org/10.1371/journal.pone.0172762
Number of Alzheimer's deaths found to be underreported. (May 2014). National Institute on Aging. NIH. https://www.nia.nih.gov/news/number-alzheimers-deaths-found-be-underreported
Padda IS, Parmar M. Aducanumab. (November 2022). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK573062/
Parkinson’s Disease. (n.d.) Harvard Stem Cell Institute. The President and Fellows of Harvard College. https://hsci.harvard.edu/parkinsons-disease-0
Parkinson's Disease: Hope Through Research. (n.d) National Institute of Neurological Disorders and Stroke. NIH. https://www.ninds.nih.gov/health-information/patient-caregiver-education/hope-through-research/parkinsons-disease-hope-through-research
Phase 3 Clinical Trial Targeting Lou Gehrig's Disease Gets $15.9 Million Investment From Stem Cell Agency. (July 2017). California Insititute for Regenerative Medicine. https://www.cirm.ca.gov/about-cirm/newsroom/press-releases/07202017/phase-3-clinical-trial-targeting-lou-gehrigs-disease
Reardon, S. (2023). FDA approves Alzheimer’s drug lecanemab amid safety concerns. Nature. https://www.nature.com/articles/d41586-023-00030-3
Schulte, J., & Littleton, J. T. (2011). The biological function of the Huntingtin protein and its relevance to Huntington's Disease pathology. Current Trends in Neurology, 5, 65–78. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3237673/
Shugart, J. (July 2017). C9ORF72 Throws a Wrench into DNA Repair Machinery. Alzforum. FBRI, LLC. https://www.alzforum.org/news/research-news/c9orf72-throws-wrench-dna-repair-machinery
Sun, X., Chen, W. D., & Wang, Y. D. (2015). β-Amyloid: the key peptide in the pathogenesis of Alzheimer's disease. Frontiers in Pharmacology, 6, 221. https://doi.org/10.3389/fphar.2015.00221
The Ionis antisense pipeline. (n.d.) Ionis Pharmaceuticals, Inc. https://www.ionispharma.com/ionis-innovation/pipeline/
van Dyck, C. H., Swanson, C. J., Aisen, P., Bateman, R. J., Chen, C., Gee, M., Kanekiyo, M., Li, D., Reyderman, L., Cohen, S., Froelich, L., Katayama, S., Sabbagh, M., Vellas, B., Watson, D., Dhadda, S., Irizarry, M., Kramer, L. D., & Iwatsubo, T. (2023). Lecanemab in Early Alzheimer's Disease. The New England journal of medicine, 388(1), 9–21. https://doi.org/10.1056/NEJMoa2212948
van Rheenen, W., Shatunov, A., Dekker, A. et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48, 1043–1048. https://doi.org/10.1038/ng.3622
Xu, J. (September 2015). QuickStats: Age-Adjusted Death Rates for Parkinson Disease — United States, 2000–2013. Morbidity and Mortality Weekly Report. National Vital Statistics System, 64 (36);1034. Mortality public use data files, 2000–2013. Centers for Disease Control and Prevention. https://www.cdc.gov/mmWr/preview/mmwrhtml/mm6436a9.htm
What are the Signs of Alzheimer’s Disease? (2017). National Institute on Aging. NIH. https://www.nia.nih.gov/health/what-are-signs-alzheimers-disease
What is ALS? (n.d.) The ALS Association. https://www.als.org/understanding-als/what-is-als

.jpg)