The Search For New Ways To Intervene in Alzheimer’s Disease
- Published16 Dec 2021
- Author Jennifer Michalowski
- Source BrainFacts/SfN
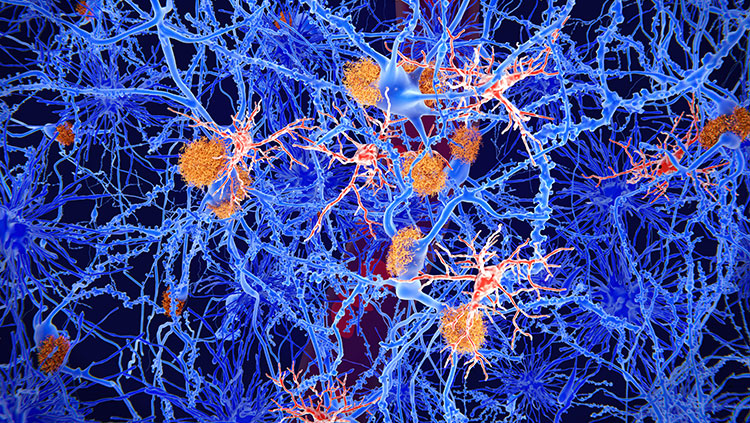
In 1906, German psychiatrist Alois Alzheimer examined the brain of a deceased patient under a microscope and found abnormal deposits in and around the cells. He linked the plaques and tangles he observed to the disorientation, memory loss, and hallucinations that this patient experienced before death. Those distinctive deposits remain the defining features of Alzheimer’s disease. It took nearly 80 years to begin to tease apart the origins of those deposits and their relationship to patients’ symptoms — an effort that gained momentum when researchers identified the plaques between brain cells as clumps of a protein called amyloid-beta.
One of those researchers, geneticist John Hardy, chair of the molecular biology of neurological disease department at the University College London’s Dementia Research Institute, discovered mutations in the amyloid gene in two families with Alzheimer’s disease. The amyloid protein became a suspected culprit in the disease and a primary focus of drug developers’ attention. Not long after, the tangle-forming tau protein joined the list of suspects.
In 2018, the Lundbeck Foundation awarded its prestigious Brain Prize to Hardy and three other researchers, Bart De Strooper, Michel Goedert, and Christian Haass, for their research on the genetic and molecular basis of Alzheimer’s disease.
Identifying the targets for treatment
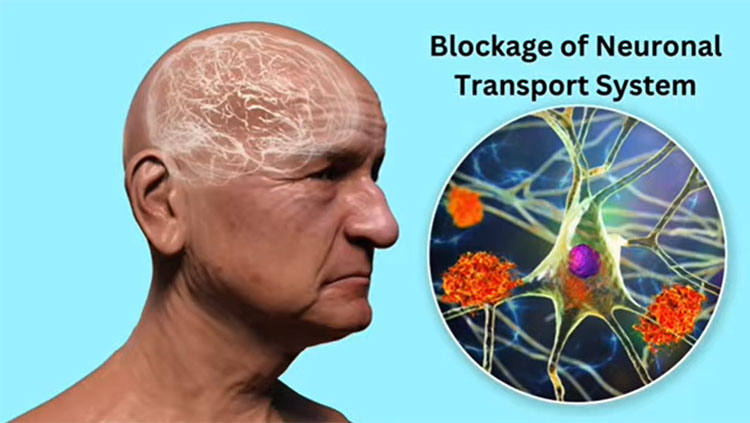
The brains of people with Alzheimer’s disease undergo profound cell loss, particularly in parts of the brain involved in memory. As Alzheimer observed early in the 20th century, plaques of what is now known to be amyloid-beta collect in the spaces between cells. In later stages of the disease, protein tangles are visible inside cells, and these cells show visible damage. In 1988, Michel Goedert and colleagues determined that the tangled filaments inside neurons are made of a protein called tau.
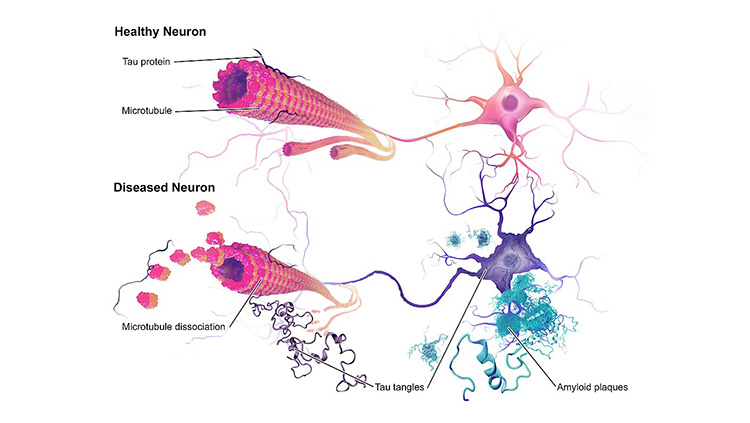
“The problem we had had from Alzheimer's time is knowing what element of that pathology came first,” Hardy says. In the 1980s, Hardy’s team found mutations in the gene that encodes amyloid in families affected by a rare genetic form of the disease. “That told us in that family, that amyloid was where the disease started. So, it started to put an order to the pathology,” he says. Haass and De Strooper’s labs helped reveal how plaque-forming amyloid-beta is generated from a larger precursor protein produced in healthy cells.
Genetics also indicated that the tangles of tau protein that form inside neurons contribute to patients’ symptoms. Several groups showed in the late 1990s that people with mutations in the gene for tau developed dementia, even in the absence of amyloid plaques.
“[The identification of amyloid mutations in families affected by the disease] really led to the idea that if we want to treat, especially early in the disease, we need to be treating where the disease starts,” Hardy says. “Since then, there's been an increasing push to develop therapies aimed at amyloid.”
The future of Alzheimer’s treatment
In June 2021, the U.S. Food and Drug Administration granted accelerated approval to aducanumab (Aduhelm), a therapy designed to clear amyloid plaques from the brain. That approval was controversial, and the drug failed to garner a recommendation from the FDA’s advisory committee. Hardy says he expects aducanumab, which is the first approved amyloid-targeted therapy, will have a “marginally beneficial effect” for a subset of patients whose Alzheimer’s disease symptoms are mild.
Tau tangles develop later in the course of the disease, and Hardy says their presence correlates more closely to the severity of patients’ dementia than amyloid plaques. “We know that tangle pathology on its own can lead to dementia, and that can be caused by tau mutations,” he says. “So that is definitely a target for sure — perhaps a little later in the disease process.” Therapies that aim to prevent or eliminate tau tangles are in various stages of development.
Hardy says drug developers need to pursue multiple strategies to reduce the burden of Alzheimer’s disease. Patients might even benefit from combinations of therapies that target different aspects of Alzheimer’s pathology.
“We need better amyloid drugs, for sure. Early in the disease, we target amyloid, but I think we also need to look at other targets too,” he says. “We are looking for tau drugs, of course, and some of those are just reaching the clinic. That's a very good target.”
There’s a third strategy that Hardy and others are investigating, as well. Researchers have learned inflammation triggered by microglia, immune cells in the brain, is also important to the disease’s progression. “We know now that the brain's microglia react to the amyloid deposition, and we know that genetic variability in how they react is important in the disease,” Hardy says. “So I think another therapeutic target is understanding and improving that reaction of microglia to amyloid deposition.” By exploring how and why microglia’s response to amyloid varies across individuals, researchers may be able to identify new ways to intervene.
“The next generation [of scientists] is going to be the generation which makes a real difference in dementia,” Hardy says. With the tools of modern biology and knowledge gleaned from decades of research, “we’re not shooting in the dark anymore.”
About the Author

Jennifer Michalowski writes about scientists and their discoveries, exploring topics ranging from ancient evolution to the newest insights about the brain.
CONTENT PROVIDED BY
BrainFacts/SfN
References
Glenner, G. G., & Wong, C. W. (1984). Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications, 120(3), 885–890. https://doi.org/10.1016/s0006-291x(84)80190-4
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving, N., & James, L. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature, 349(6311), 704–706. https://doi.org/10.1038/349704a0
Goedert, M., Wischik, C. M., Crowther, R. A., Walker, J. E., & Klug, A. (1988). Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proceedings of the National Academy of Sciences of the United States of America, 85(11), 4051–4055. https://doi.org/10.1073/pnas.85.11.4051