Disease-Causing Proteins
- Published24 Apr 2013
- Reviewed24 Apr 2013
- Author Aalok Mehta
- Source BrainFacts/SfN

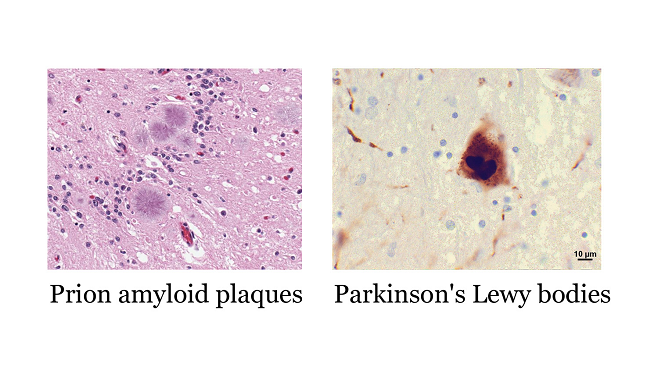
Understanding the cause of “mad cow” disease and related conditions resulted in a fundamental shift in what scientists understood about proteins in brain disease.
Searching for the cause of a rare group of brain diseases revolutionized what scientists know about brain proteins — especially the surprisingly common and complex ways in which they can act in harmful ways. This new understanding, which was made possible by the funding of basic science, may someday lead to improved insights and possibly better treatments for brain diseases in which proteins tend to misfold, including Alzheimer's, Parkinson’s, and Huntington's diseases.
Rare Disorders Traced to Proteins
A group of rare brain diseases may seem like an unlikely place to turn for clues about the estimated 36 million cases of Alzheimer's disease and related dementias worldwide. But, the shocking discovery that a misfolded protein was behind the mysterious disorders known as transmissible spongiform encephalopathies (TSEs) forever changed the way scientists think about proteins and their roles in many neurodegenerative diseases.
The Mystery Behind Rare Neurological Disorders
TSEs strike people as well as animals. Creutzfeldt-Jakob disease (CJD) — a rare dementia that affects about one in every one million people each year — is one of the most well-known of the human TSEs. Bovine spongiform encephalopathy (BSE), or “mad cow” disease, and scrapie, a neurological disease that strikes sheep and goats, are also TSEs.
In 1972, after meeting a patient with CJD, neurology resident Stanley Prusiner became determined to uncover the source of the disease. Because studies suggested CJD could be transmitted, many scientists believed a small virus was to blame.
Misfolded Protein Leads to Infection
After a decade of research funded by the National Institutes of Health, Prusiner successfully isolated the agent in question. What he found surprised him: CJD traced back to a single protein. Prusiner named this protein "prion" (combining “protein” and “infectious”).
Scientists were shocked. How could a protein, which lacks genetic instructions, result in changes that cause disease? Many scientists questioned Prusiner’s findings, suggesting instead he had overlooked the true source of the mysterious diseases.
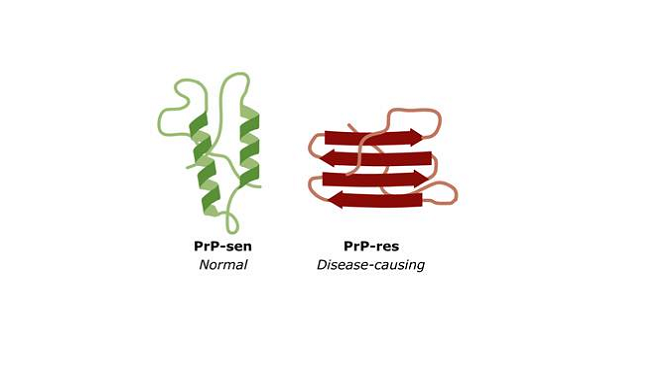
Despite immense skepticism, Prusiner kept at it. In 1984, he and his colleagues shocked scientists again with the discovery that a form of prion protein (PrP) was present in healthy people and animals. As it turns out, prion proteins can fold into two distinct shapes: one is harmless; the other leads to disease (like CJD). Remarkably, the misfolded, disease-causing prions have the ability to "corrupt" the harmless versions, causing them to misfold as well. For this work, Prusiner won the 1997 Nobel Prize in Physiology or Medicine.
Prions Play Leading Role in Mad Cow Outbreak
Just as Prusiner's ideas about prions were beginning to gain traction in the late 1980s, pockets of cattle in the United Kingdom began displaying aggressive behavior, weight loss, and difficulties standing — symptoms indicative of the neurodegenerative illness that would later come to be called mad cow disease.
Mad Cow Traced to Prions
The affected cattle, as it turns out, had consumed prion-infected feed. By the time the animals showed signs of the disease, the infectious prion proteins had been silently corrupting healthy proteins in the brain for some time. As the numbers of healthy proteins converted to the misfolded, disease-causing form, the areas of the brain where the misfolded proteins accumulated began to deteriorate. Scientists believe the mad cow outbreak was amplified and spread by the inclusion of affected animal tissue in the feed offered to healthy animals.
With no cure or ability to detect prions in living people or animals, stopping BSE required extreme steps. Government authorities banned using cattle tissue in feed and ordered the slaughter of older cattle to limit the spread of the disease.
Disease Moves from Animal to Human
Despite significant efforts to contain BSE, some people unknowingly consumed meat from the infected cattle. Research suggests the consumption of beef from cattle with BSE may have led to the emergence of a disease similar to CJD called variant CJD (vCJD) that began striking and killing younger people in the United Kingdom in 1996.
The misfolded prion proteins present in vCJD as well as in other TSEs tend to cluster into thick, compact sheets that are highly resistant to heat, the body's immune system, and even protein-busting enzymes. Clumps of the abnormal proteins can grow in the brain for years without notice. Eventually, this buildup blocks the ability of nerve cells to communicate and causes cell death. Research on the infectious form of prion proteins in prion diseases led scientists to look closely at the role of misfolded proteins in other neurodegenerative diseases.
Future Treatments for Neurodegenerative Disease
Prion diseases aren't the only diseases in which misfolded proteins result in toxic clumps in the brain. In Alzheimer's, Parkinson's, and Huntington's diseases, plaques and tangles made up of different types of protein also build up, leading to nerve cell death.
New research suggests that the proteins in these diseases, while different than prions, share similarities with the misfolded, disease-causing proteins. For example, some animal studies show that the beta-amyloid protein, which is associated with Alzheimer's disease, can corrupt other proteins and cause new protein plaques to form when injected into the brain.
Scientists hope that efforts to uncover how disease-causing proteins corrupt healthy proteins in prion diseases and Alzheimer’s disease will inform future treatments for slowing the progression of neurodegenerative conditions.
Mystery Propels More Research
Even after decades of research, prions remain highly mysterious. Scientists continue to search for answers as to why some prion diseases arise spontaneously and the mechanism the misfolded proteins use to induce healthy cellular proteins into the misfolded, disease-causing form.
Scientists know that normal, healthy prion protein (PrP) has an important function, but specific information about its role remains elusive. Mice bred to lack PrP are immune to prion diseases, but show only subtle neurological changes, for instance. Recent evidence suggests that PrP may help protect nerve cells during stress or disease, and assist with the formation of new connections, or synapses, between brain cells, which are essential to learning and memory.
When Prusiner first set out to identify the agent responsible for the rare, but deadly prion diseases, little did he know his findings would have health implications far beyond, as is often the case with basic research. Sustained funding from NIH, the National Science Foundation, and the U.S. Department of Agriculture continues to be vital to scientists’ quest to understand the events that lead to protein misfolding in the brain and develop new therapies for the millions of people worldwide suffering from neurodegenerative diseases.
CONTENT PROVIDED BY
BrainFacts/SfN
References
Belay ED, Schonberger LB. The public health impact of prion diseases. Annual Review of Public Health. 26, 191-212 (2005).
Chung E, Prelli F, Dealler S, Lee WS, Chang, Y-T, et al. Styryl-based and tricyclic compounds as potential anti-prion agents. PLoS ONE. 6(9) (2011).
Eisele YS, Obermüller U, Heilbronner G, et al. Peripherally applied aβ-containing inoculates induce cerebral β-amyloidosis. Science. 330:980-982 (2010).
Gibbons RA, Hunter GD. Nature of the scrapie agent. Nature. 215:1041-1043 (1967).
Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. Journal of Neurochemistry. 95: 1373-1386 (2005).
Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 216: 136-44 (1982).
Prusiner SB. Molecular biology of prion diseases. Science. 252:1515-22 (1991).
Prusiner SB. Prion diseases and the BSE crisis. Science. 278:245-51 (1997).
Si K, Lindquist S, Kandel ER. A neuronal isoform of the Aplysia CPEB has prion-like properties. Cell. 115: 879 - 891 (2003).